International Journal of Anatomical Sciences 2013, 4(1):27-28
Research Paper
Study of Holoprosencephaly
Melani Rajendran S, Venkatasai PM.
Dept. of Anatomy, Matha Dental College and Hospital, Chennai – 600 069, Tamil Nadu, India,
Dept. of Radiology and Imaging Sciences, Sri Ramachandra Medical College and Research Institute, Chennai – 600 116, Tamil Nadu, India.
Key words: holoprosencephaly, alobar holoprosencephaly, semilobar holoprosencephaly, lobar holoprosencephaly, neural tube defect
Abstract: Holoprosencephaly is a rare congenital brain malformation that occurs during development due to incomplete separation of the cerebral hemispheres .It may be caused due to hereditary factors, chromosomal anomalies, environmental teratogenic factors. Four cases of holoprosencephaly were observed during a period of one year and their features are discussed.
Neonatal brain injury is very important since it predicts subsequent infant mortality and morbidity in the premature infants. The term holoprosencephaly is proposed by De Myer (1963). Holoprosencephaly is a disorder resulting from failure of septation, cleavage, or differentiation of the midline forebrain structures at various levels or to various degrees (De Myer, 1963).It is caused by a primary defect in patterning and induction of the basal forebrain during embryogenesis, causing the improper development of the brain and resulting in incomplete division of the cerebral hemispheres (Stashinkio, 2004). Holopros-encephaly most commonly affects the telencephalon and the diencephalon (De Myer, et al. 1963). According to the degrees of failed differentiation of the forebrain, the holoprosencephaly is classified into 4 categories: alobar holoprosencephaly, where the brain is not divided, semilobar holoprosencephaly in which the cerebral hemispheres is incompletely developed and lobar holoprosencephaly in which the brain has somewhat divided which is the least severe form. Syntelencephaly, or middle interhemispheric variant of holoprosen-cephaly (Armand Marie Leroi, 2003), in which the posterior parts of the frontal and parietal lobes are improperly separated, but the rostro-basal forebrain properly separa-tes; it is also currently classified as a type of holoprosencephaly (Armand Marie Leroi, 2003).
Holoprosencephaly is associated with facial anomalies including cyclopia,proboscis, ethmocephaly or cebocephaly (Kinsman, et al. 2000), microcephaly, midface flattening, hypotelorism, flat nasal bridge and single maxillary central incisor, premaxilla agenesis, median cleft palate and cleft lip and other less-severe facial anomalies (Wallis and Merenke, 2000).
The aim of this study is to increase the understanding and awareness of genetic and clinical manifestations of holoprosen-cephaly. Therefore a study was carried out to find out the probabilities of occurrence of holoprosencephaly among the neural tube defects for a period of one year. 40 cases of neural tube defects were found, among which 4 cases of holoprocencephaly were identified and reported.
Materials and Methods
About 1500 antenatal scans were done for a period of one year, in the I, II and III trimester; out of which, 40 central nervous system anomalies were found and in turn, 4 of them showed holoprocencephaly.
They were:
Alobar prosencephaly – 2
Semilobar prosencephaly – 1
Lobar prosencephaly – 1
Autopsy was done with the concern of the parents. Autopsy findings were correlated with sonographic findings. CNS anomalies not compatible with life were terminated
Observations
Alobar holoprosencephaly
The foetuses were 19 weeks & 20 weeks old respectively They were found with alobar holoprosencephaly (Figs.1a &1b). The parents of both the foetuses were consanguineous and one of the parents suffered from infertility for 5 years. One of the fetuses had cyclops and with proboscis. Their brains showed no separation of cerebral hemispheres; absence of falx cerebri; a large single central ventricle; thin and fused thalami; small third ventricle and the midbrain with a small cerebral aqueduct.
Semilobar holoprosencephaly
The foetus was 22 week old with semilobar holoprosencephaly (Figs.2a & 2b). Its parents were non consanguineous. The foetus was found with median cleft lip. It had a small single central ventricle; fused frontal and parietal lobes bilaterally; inter hemispheric fissure was present only posteriorly; Lack of cleavage of basal ganglia and thalami; absence of body of corpus callosum but genu and splenium were present.
Lobar holoprosencephaly
The foetus was 20 week old with lobar holoprosencephaly The parents were non consanguineous. The foetus had median cleft lip and cleft palate. The brain was found with right and left cerebral hemispheres but the frontal lobes were fused and the lateral ventricles were separated (Figs.3a,b & 3c).
Discussion
The holoprosencephaly is a rare congenital malformation and one of the neural tube defects. It is classified into 4 categories: alobar holoprocencephaly, in which nondivision of the brain and with associated facial anomalies. The brain show absence of the interhemispheric fissure, falx cerebri, third ventricle and fused thalami and often absence of neurohypophysis and olfactory tracts (De Myer et al., 1963); semilobar holoprocencephaly in which cerebral hemispheres is incompletely developed with posterior partial formation of the interhemispheric fissure, with only a single ventricle, variant heterotopic gray matter ; lobar holoprosencephaly is the least severe form in which the brain has somewhat divided. The brains are with the presence of an interhemispheric fissure but the cingulate gyrus and fused lateral ventricle and absence of septum pellucidum (De Myer et al., 1963) and syntelencephaly, or middle interhemispheric variant of holoprosen-cephaly (MIHV), in which the posterior frontal and the parietal lobes are not improperly separated, but the rostro-basal forebrain properly separates. Earlier it is not considered as a variant of HPE at all, but is currently classified as a type of holoprosencephaly (Armand Marie Leori, 2003).
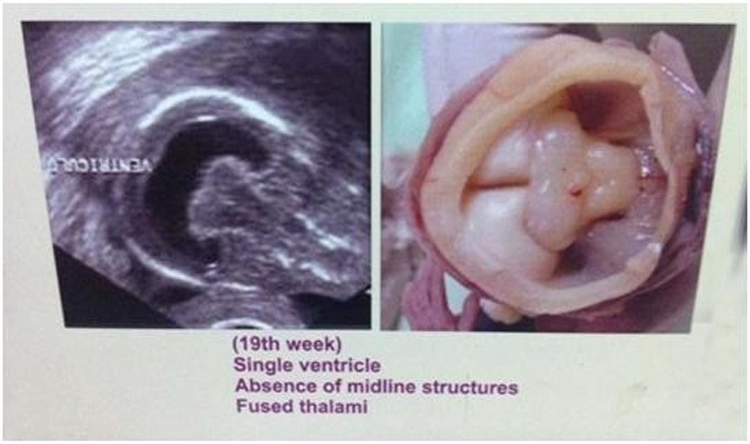 |
Fig 1a & b Alobar holoprosencephaly
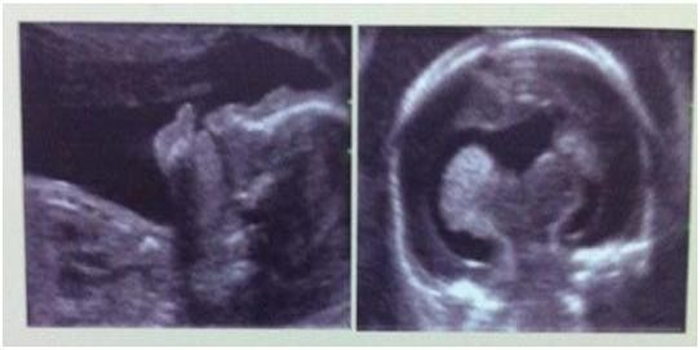 |
Fig. 2a & b Semilobar holoprosencephaly (22nd week of pregnancy)
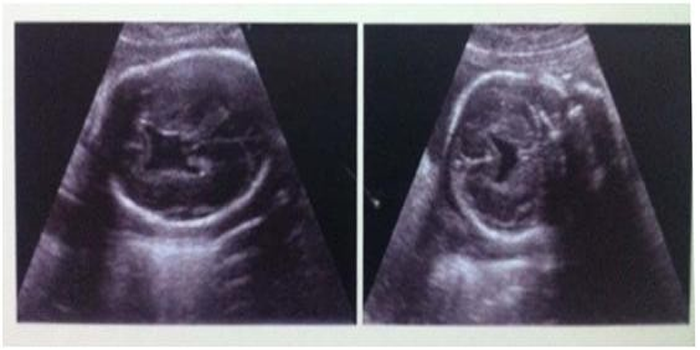 |
Fig. 3a & b Lobar holoprosencephaly
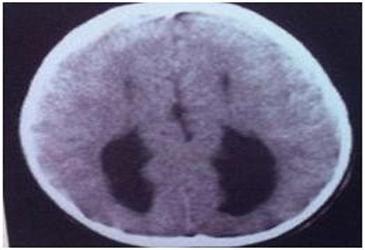 |
Fig. 3c Lobar prosencephaly
The four cases of the holoprocen-cephaly observed in the present study also showed majority of the malformations reported above.
Apart from the facial anomalies, associated abnormalities include micro-cephaly, hydrocephalus, agenesis of the corpus callosum, posterior cranial fossa abnormality, cerebellar vermis aplasia, myelomeningocele, absence of an olfactory bulb, hypothalamic and brainstem dysfunc-tion, increased muscle tone, sleep disturbances (Barr and Cohen, 1999), renal dysplasia, renal cysts, cardiovascular malformations, intestinal abnormalities, omphalocele, gastro oesophageal reflux, choking etc, club foot, sirenomelia,(Chen et al. 1997), spina bifida, and endocrinopathies (pituitary gland dysplasia, growth hormone deficiency and diabetes insipidus (Traggiai and Stanhope, 2002), polydactyly (Verloes et al. 1991), inability to smell, develop-mental delay, intellectual impairment, seizures etc. (Bronshtein and Wiener, 1991).
Development
During the third week of embryonic life, the prechordal mesoderm migrates into the area prior to the notochord and affects midline facial development. Hence before 4 weeks of embryonic age, the varying degrees of loss or disruption in the development of prechordal mesoderm cause abnormal forebrain development, midfacial defects as well as fusion of lateral ventricles and the 3rd ventricle (Muenke and Beachy, 2000). Division of cerebrum into right and left halves normally occurs at the end of the 5th or the beginning of 6th week of gestation.
Birth prevalence rate
During early embryogenesis, the prevalence rate is about 1 in 250; s the prevalence rate in live births ranges from 1: 14,736 to 1: 26,730 due to the high rate of spontaneous abortion, the prevalence rate in live births ranges from (Cohen,1989); Bullen et al. (2001) represented the total prevalence (including pregnancy termination) was 1.2 cases per 10,000 registered births, and the birth prevalence (affected live births and stillbirths at > 24 weeks’ gestation) was 0.49 cases per 10,000 births. But according to Dubourg et al. 2007, holo procencephaly is seen in 1 per 10,000 to 16,000 live births. In most cases of holoprosencephaly, if the malformations are so severe the babies die before birth (Tatori et al. 2005).
Etiology
Knowledge of the etiologies of holoprosencephaly is important for establishing the risk of recurrence. The etiology of holoprosencephaly indicates interactions with both genetic and environmental factors which include chromosomal anomalies, gene rearrange-ments, Mendelian mutations and teratogens.
Geneticfactors
The majority of holoprosencephaly cases are autosomal dominant, autosomal recessive, or X-linked in inheritance. Almost 50% of all holoprosencephaly cases havecytogenetic abnormalities and approximately 18% -25% of patients of holoprosencephaly have a documented monogenic syndrome (Croen et al. 1996; Olsen et al. 1997). Approximately half of all infants or fetuses with holoprosencephaly also have chromosomal abnormalities, most often trisomy 13 (Cohen, 1989; Mcgahan et al. 1990; Muenke, 1994; Rasmussen et al. 1996; Ming et al. 1998; Bullen et al. 2001; Blaas et al. 2002).
Environmental factors
Environmental factors are mainly maternal diabetes mellitus which increase holoprosencephaly risk (Ramos-Arroyo et al. 1992; Muenke, 1994; Ming and Mueuke, 1998; Peebles, 1998; Croen, 2000; steroid alkaloids, thanol, and retinoic acid (Sulik et al. 1995). X-ray exposure, maternal alcohol consumption during early pregnancy (Cohen, 1989; 2002), steroid alkaloids, thanol and retinoic acid (Sulik et al. 1995). X-ray exposure, maternal alcohol consumption. Other factors which have teratogenic effects are maternal smoking, respiratory illness medications and salicylate-containing medications, estrogen/progestin, anticonvulsants, weight reduction diets and low maternal weight, previous pregnancy loss and congenital infection with cytomegalovirus, rubella, and toxoplasmosis (Croen et al. 1996). Women with early menarche are more likely to have holoprosencephaly (Croen et al. 2000).
Conclusion
Because of the short life span and ominous outcome in all patients with holoprosencephaly, genetic counseling and prenatal diagnosis is essential. The earliest gestational age at the time of diagnosis is 14 weeks (Bronshtein and Wiener, 1990). Prenatal ultrasound can detect the more severe forms of holoprosencephaly and associated defects such as hydrocephaly (Chervenak, 1985; Vintzileos et al. 1987; Peebles,1998).Therefore prenatal diagnosis and with the consent of the parents, elective termination reduce the birth prevalence of holoprosencephaly (Croen, 1996; Forrester, 2000; Bullen, 2001; Blaas, 2002). Knowledge of the different varieties of holoprocencephaly helps the clinician to arrive at an antenatal diagnosis and to decide about the fetal outcome.
References
Armand Marie Leroi (2003) Mutants: On the Form, Varieties and Errors of the Human Body, Harper Perennial, London. ISBN 0-00-653164-4.
Barr M Jr, Cohen MM Jr (1999) Holoprosencephaly survival and performance. Am J Med Genet, 89:116-20.
Blaas HG, Eriksson AG, Salvesen KA, Isaksen CV, Christensen B, Mollerlokken G, Eik-Nes SH (2002) Brains and faces in holoprosencephaly: pre- and postnatal description of 30 cases. Ultrasound Obstet Gynecol, 19:24-38.
Bronshtein M, Wiener Z (1991) Early transvaginal sonographic diagnosis of alobar holoprosencephaly. Prenat Diagn, 11:459-64.
Bullen PJ, Rankin JM, Robson SC (2001) Investigation of the epidemiology and prenatal diagnosis of holoprosencephaly in the North of England. Am J Obstet Gynecol, 184:1256-1262.
Chen CP, Shih SL, Liu FF, Jan SW(1997) Cebocephaly, alobar holoprosencephaly, spina bifida, and sirenomelia in a stillbirth. J Med Genet, 34:252-5.
Chervenak FA, Isaacson G, Hobbins JC, Chitkara U, Tortora M, Berkowitz RL (1985) Diagnosis and managment of fetal holoprosencephaly. Obstet Gynecol, 66:322-326.
Cohen MM (1989) Perspectives on holoprosencephaly: Part 1. Epidemiology, genetics, and syndromology. Teratology, 40:211-235.
Cohen, M, Shiota, K (2002). Teratogenesis of Holoprosencephaly. Amercian Journal of Medical Genetics, 109:1-15.
Czeizel A (2004) The primary prevention of birth defects: multivitamins or folic acid? International Journal of Medical Sciences, 1:1:50-54.
DeMyer W, Zeman W, Palmer CG (1963) The face predicts the brain: diagnostic significance of median facial anomalies for holoprosencephaly (arhinencephaly). Pediatrics; 34:256-63.
Forrester MB, Merz RD (2000) Epidemiology of holoprosencephaly in Hawaii, 1986-97. Ped Perinatal Epidemiol, 14:61-63.
Kinsman S, Plawner L, Hahn J (2002) Holoprosencephaly: recent advances and new insights. Current Opinion in Neurology, 13: 127-132.
Mcgahan JP, Nyberg DA, Mack LA (1990) Sonography of facial features of alobar and semilobar holoprosencephaly. AJR Am J Roentgenol, 154 (1): 143-8.
Ming JE, Muenke M (1998) Holoprosencephaly: from Homer to Hedgehog. Clin Genet, 53:155-163.
Muenke M (1994) Holoprosencephaly as a genetic model to study normal craniofacial development. Semin Dev Biol, 5:293-301
Odent S, Le Marec B, Munnich A, Le Merrer M, Bonaiti-Pellie C (1998) Segregation analysis in nonsyndromic holoprosencephaly. Am J Med Genet,77:139-143.
Olsen CL, Hughes JP, Youngblood LG, Sharpe-Stimac (1997) Epidemiology of holoprosencephaly and phenotypic characteristics of affected children: New York State, 1984-1989. Am J Med Gen, 73:217-226.
Peebles DM (1998) Holoprosencephaly. Prenat Diagn , 18:477-480.
Ramos-Arroyo MA, Rodriguez-Pinilla E, Cordero JF (1992) Maternal diabetes: the risk for specific birth defects. Eur J Epidemiol, 8:503-508.
Rasmussen SA, Moore CA, Khoury MJ, Cordero JF (1996) Descriptive epidemiology of holoprosencephaly and arhinencephaly in Metropoitan Atlanta, 1968-1992. Am J Med Gen, 66:320-333.
Stashinkio, E, Clegg, N, Kammann, H, Sweet, V, Delgado, M, Hahn, J, Levey, E (2004) A retrospective survey of perinatal risk factors of 104 living children with holoprosencephaly. American Journal of Medical Genetics, 128A:114-119.
Sulik KK, Dehart DB, Rogers JM, Chernoff N (1995) Teratogenicity of low dosed of all-trans retinoic acid in persomite mouse embryos. Teratology, 51:398-403.
Totori-Donati, Paolo; Rossi, Andrea; Biancheri, Roberta (2005) “Brain Malformations”. In Totori-Donati, Paolo; Rossi, Andrea; Raybaud, C. Pediatric Neuroradiology: Brain, Head, Neck and Spine 1. Springer. pp. 92–95. ISBN 3-540-41077-5.
Traggiai C, Stanhope R (2002) Endocrinopathies associated with midline cerebral and cranial malformations. J Pediatr, 140:252-5.
Verloes A, Ayme S, Gambarelli D, Gonzales M, Le Merrer M, Mulliez N, Philip N (1991) Holoprosencephaly – polydactyly (pseudotrisomy 13′) syndrome: a syndrome with features of hydrocephalus and Smith-Lemli-Opitz syndrome. A collaborative multicentre study. J Med Genet, 28:297-303.
Vintzileos AM, Campbel WA, Nochimson DJ, Weinbaum PJ (1987) Antenatal evaluation and management of ultrasonically detected fetal anomalies. Obstet Gynecol, 69:640-660.
Wallis D, Muenke M (2000) Mutations in holoprosencephaly. Hum Mutat, 16:99-108.